



Первые клинические испытания на людях – FIH CTs (Part I)
- 12 апреля 2017
- 1 фаза, FIH, IMP, замена, интегрированный протокол клинического исследования, исследуемый препарат, концепция 3R, начальные клинические испытания, первые исследования на людях, первые клинические испытания на людях, снижение риска, сокращение, усовершенствование,

Clinical trials are essential for the development of medicines and without them patients cannot gain access to new potentially life-saving medicines. EU and international guidelines are in place to ensure that first-in-human clinical trials are conducted as safely as possible. EMA’s existing guideline, released in 2007, provides advice on first-in-human clinical trials, in particular on the data needed to enable their appropriate design and allow the initiation of treatment in trial participants.
Between July and end of September 2016, EMA released for public consultation a concept paper which outlined the major areas that needed to be revised in the guideline, to reflect the evolution of practices in the last ten years. The review also took into account the lessons learnt from the tragic incident which took place during a phase I first-in-human clinical trial in Rennes, France, in January 2016.
Currently, the European Medicines Agency (EMA), in cooperation with the European Commission and the Member States of the European Union (EU), is proposing changes to its existing guideline on first-in-human clinical trials, to further improve the safety of trial participants, whether patients or healthy individuals. The revised guideline was open for public consultation until 28 February 2017.
The revision is intended to assist sponsors in the transition from non-clinical to early clinical development and identifies factors influencing risk for new investigational medicinal products (IMPs). The document includes considerations on quality aspects, non-clinical and clinical testing strategies and designs for first-in-human (FIH) clinical trials (CTs) and early phase CTs. Strategies for mitigating and managing risks are given, including principles on the calculation of the starting dose to be used in humans, the subsequent dose escalation, the criteria for maximum dose and the conduct of the CT including the conduct of multiple parts.


This revised guideline aims to address the increasing complexity of protocols of first-in-human clinical trials, i.e. integrated protocols that combine a number of different study parts (e.g. single ascending dose (SAD), multiple ascending dose (MAD), food effects, or different age groups), in recent years.
The guideline applies to all new chemical and biological IMPs.
It should be underlined here that careful dosing selection of an IMP is a vital element to safeguard the subjects participating in FIH and early CTs.
All available non-clinical information (PD, PK, TK and toxicological profiles, etc.) should be taken into consideration for the calculation of the starting dose, dose escalation steps and maximum dose. Non-clinical regulatory testing of medicinal products for human use is carried out to support clinical trials and marketing authorisation (MA) applications. For both human and veterinary medicinal products regulatory testing is also conducted to control quality during (in-process) and/or at the end (final product batch testing) of the production of the product. In accordance with the 3Rs principles on animal use (Directive 2010/63/EU), a scientifically satisfactory method or testing strategy, not entailing the use of live animals should be used wherever possible. The use of in vitro studies, including studies using human material, is encouraged whenever possible. A general overview on implementation of 3Rs principles in this context is provided in the Guideline on the principles of regulatory acceptance of 3Rs (replacement, reduction, refinement) testing approaches. Further information on current or future implementation of specific 3Rs testing approaches for human and veterinary medicinal products can be found in separate reflection papers providing an overview of the current regulatory testing requirements for medicinal products for human and veterinary use and opportunities for implementation of the 3Rs.
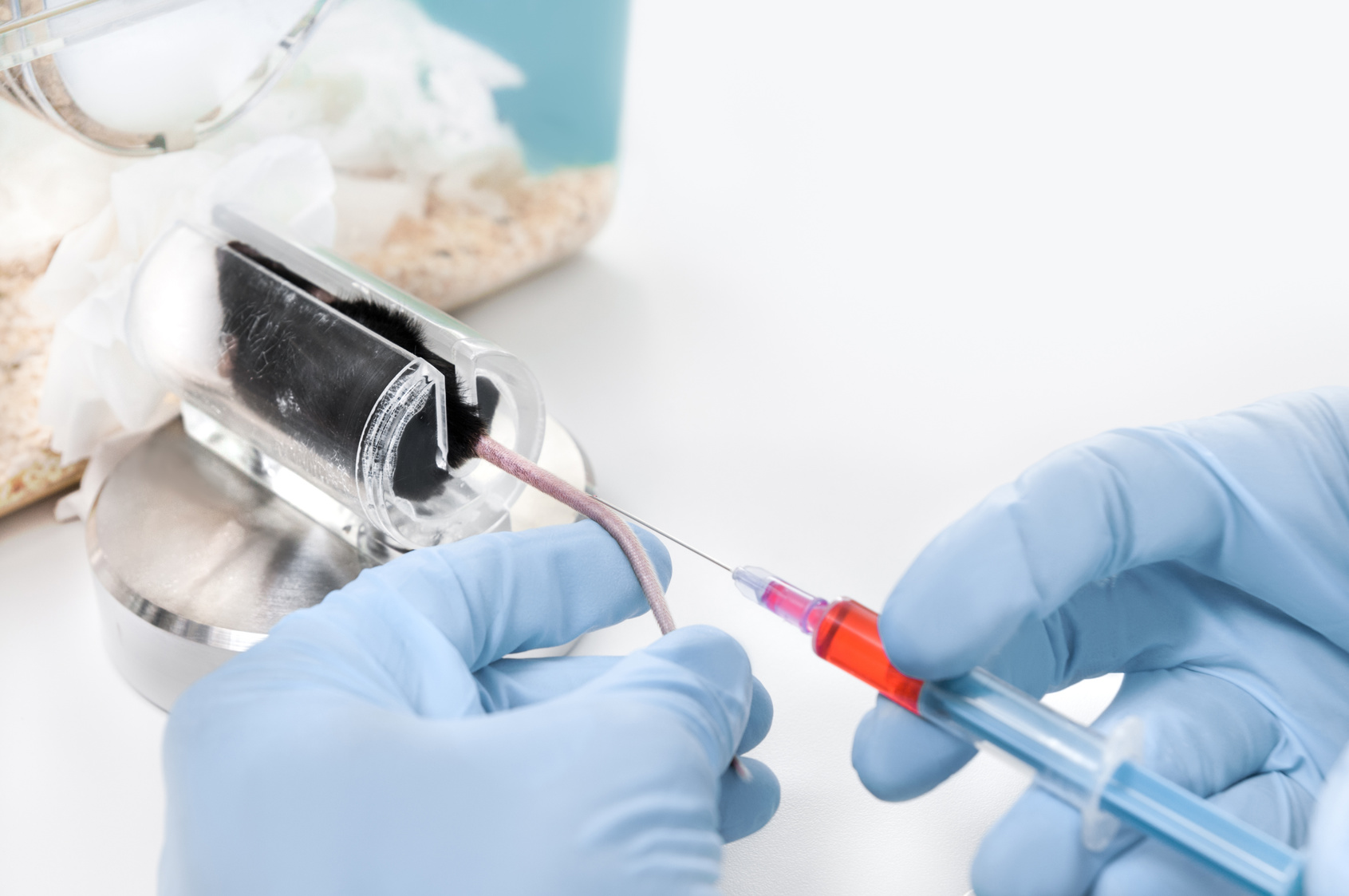

Furthermore, clinical data (e.g. PK, PD and reports of adverse events) emerging during the trial from previous dosed cohorts/individuals needs to be taken into account, in line with pre-specified decision criteria. Experience, both non-clinical and clinical, with molecules having a similar mode of action can also be useful.
If emerging clinical data reveal significant differences from non-clinical or modelling and simulation data, a substantial amendment may be required to adjust planned dose levels unless this possibility was discussed including predefined decision criteria and approved in the protocol.
A maximum dose or exposure, which should not be exceeded in the study without approval of a substantial amendment, should be pre-defined and justified in the protocol for the full CT and/or each study part.
For integrated protocols, where it may not be possible to predefine definite doses in all study parts, a clear statement should be included that the doses will be chosen based on predefined criteria. These criteria should integrate data from previous study parts once these are completed and should not exceed the maximum exposure unless justified by the sponsor when requesting a substantial amendment.
There are following recommendations regarding the time sequence for the conduct of different study parts:
- A certain overlap of SAD and MAD parts may be considered acceptable. However, any overlap should be scientifically justified and supported by a decision-tree and a review of the available data before deciding on starting the MAD part.
- Other single dose parts (e.g. food interaction (FI)) could be conducted in parallel to the SAD part provided the dose chosen and the expected exposure are equal to or lower than that which was reached in a concluded preceding SAD cohort where all relevant data has been reviewed and no dose escalation stopping criteria were met.
- Other study parts that involve multiple dosing (e.g. FI and drug-drug interaction) should not overlap with any earlier SAD or MAD cohorts. All relevant SAD and MAD data should be reviewed before starting these parts. Deviation from this should always be justified in the protocol.
For trials or trial parts that include patients, the maximum tolerated dose (MTD) (if applicable) should be clearly defined and not be exceeded once it has been determined. A trial design using a MTD approach is considered to be unethical for healthy volunteers.

The subject safety assessments that will be routinely conducted and any additional monitoring actions should be pre-specified and justified. There should also be routine general monitoring to detect potential unexpected adverse effects that are not related to known properties of the IMP. Repeated assessments, integrating all available pharmacological, PK, PD and toxicological knowledge, and rapid processing of this information are crucial for the recognition and interpretation of developing toxicity at an early or potentially reversible stage. Follow-up of subjects should be specified within the protocol (e.g. for possible delayed adverse reactions). The sponsor should justify how safety monitoring should be extended for healthy volunteers until parameters return to within the normal range or to baseline.
The protocol should define unambiguous stopping rules which result in an immediate stop to dosing. It should further be specified in the rule if it implies a final end of dosing or a possible temporary halt with dosing re-starting after a full evaluation of available data and the approval of a substantial amendment. The submitted substantial amendment should include a justification of the proposed dosing for the continuation of the trial and details of any adjustments to the protocol including additional safety monitoring, if applicable.
Updated: 2018-11-21