


Nitrosamines in human medicines – consequences for MAHs
- 7 октября 2019
- 4-(methyl)(nitroso)amino)butanoic acid, acceptable intake, active substance, AI, analysis, API, ASMF, authorities, cancer, candesartan, carcinogen, CEP, change, chemically synthesized, CHMP, competent, confirmatory, contamination, cross-contamination, detection, DIPNA, drug product, EIPNA, EMA, evaluation, finished product, formation, generics, GMP, HCl, human, hydrochloride, ICH M7, impurity, interim, irbesartan, limit, losartan, MA, MAH, manufacturer, manufacturing, manufacturing authorisation, marketing authorisation holder, medicinal product, medicines, method, MIA, mitigate, N-nitrosodiethylamine, N-nitrosodiisopropylamine, N-nitrosodimethylamine, N-nitrosoethylisopropylamine, NDEA, NDMA, nitrosamine, NMBA, non-sartans, olmesartan, OTC, pioglitazone, process, public health, ranitidine, referral procedure, review, risk, sartans, specific ring structure, temporary, test, testing, tetrazole, valsartan, variation,



EMA’s human medicines committee (CHMP) is requesting as a matter of precaution that marketing authorisation holders (MAHs) for human medicines containing chemically synthesised active substances review their medicines for the possible presence of nitrosamines and test all products at risk. If nitrosamines are detected in any of their medicines, marketing authorisation holders must inform authorities promptly so that appropriate regulatory actions can be taken.
Update [8/01/2020]
If any MAH or manufacturer identifies that any non-chemically synthesised products, including biological products, contains nitrosamine impurities, they should inform the competent authorities immediately.
Update [4/09/2020]
Following the conclusion of the review under Article 5(3), the CHMP considered that there is also a risk of presence of nitrosamines in biological medicinal products, in particular for the biological medicines with the following risk factors:
- biologicals containing chemically synthesised fragments, where risk factors similar to chemically synthesised active substances are present;
- biologicals using processes where nitrosating reagents are deliberately added;
- biologicals packaged in certain primary packaging material, such as blister packs containing nitrocellulose.
For the above reasons the current call for review has been extended to cover also all biological medicinal products for human use.
Update [22/02/2021]
The European medicines regulatory network agreed in February 2021 an implementation plan for the outcome of the CHMP scientific review under Article 5(3). This harmonised approach includes specific measures that the network will take if nitrosamines are detected in a medicine. The Implementation plan document is available here.
IMPORTANT! Update [28/10/2020]
Having considered the knowledge acquired on the presence of nitrosamines in medicinal products since the sartans referral and taking into account the data assessed within the review under Article 5(3), in particular related to the root causes where it became clear that the root causes can be numerous, concomitant, at any stage of the production or storage of the medicinal product and cannot always be characterised, the CHMP considered that the outcome of the sartan referral should be reconsidered in light of the outcome of Art. 5(3) referral.
CHMP opinion on the impact of the Article 5(3) referral on nitrosamines in human medicinal products on the referral under article 31 of Directive 2001/83/EC for sartans medicinal products containing a tetrazole ring is expected in November 2020.
The European medicines regulatory network agreed in October 2020 an implementation plan for all recommendations on reducing the risk of impurities in medicines and ensuring that regulators are better prepared to manage cases of unexpected impurities that have been described in a report on lessons learnt from the presence of N-nitrosamines in sartan medicines. The Implementation plan document is available here.
Update [14/12/2020]
EMA’s human medicines committee (CHMP) has aligned recommendations for limiting nitrosamine impurities in sartan medicines with recent recommendations it issued for other classes of medicines.
The main change concerns the limits for nitrosamines, which previously applied to the active ingredients but will now apply instead to the finished products (e.g. tablets). These limits, based on internationally agreed standards (ICH M7(R1)), should ensure that the excess risk of cancer from nitrosamines in any sartan medicines is below 1 in 100,000 for a person taking the medicine for lifelong treatment.
In line with previous recommendations, companies should have appropriate control strategies to prevent or limit the presence of nitrosamine impurities as much as possible and, where necessary, improve their manufacturing processes. Companies should also evaluate the risk of nitrosamines being present in their medicines and carry out appropriate tests.
Update [03/03/2021]
COMMISSION IMPLEMENTING DECISION of 19.2.2021 amending Commission Decision C(2019) 2698 of 2 April 2019 concerning, in the framework of Article 31 of Directive 2001/83/EC of the European Parliament and of the Council, the marketing authorisations of medicinal products for human use which contain the active substances “candesartan”, “irbesartan”, “losartan”, “olmesartan”, “valsartan” is available here and Annexes (Annex I Scientific conclusions and Annex II Amendments to the conditions to the marketing authorisation(s)) are available here.
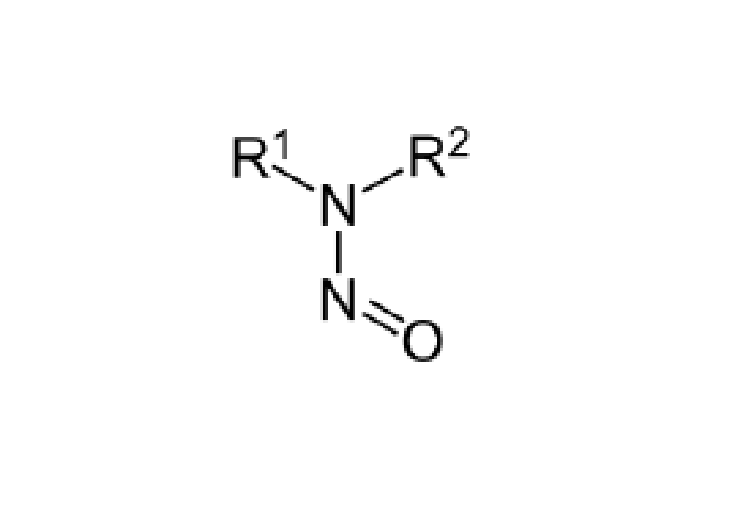
Nitrosamines are classified as probable human carcinogens, which means that long-term exposure above certain levels may increase the risk of cancer. They are present in some foods and drinking water supplies and where they have been found in medicines the risk of developing cancer has been low.
It should be reminded here that the findings of the review of sartans with a tetrazole ring indicate that there is a potential for nitrosamines to be present in APIs for other medicines (i.e. non-sartans APIs), depending on the API and the finished product manufacturing processes.
It should be also noted that traces of N-nitrosodimethylamine (NDMA) have been found in pioglitazone hydrochloride from one API manufacturer. As the nitrosamine levels in this case were within the interim limits established in the sartans review (please refer to the table below), no market action was deemed necessary. As a precaution, companies using certain reagents to manufacture pioglitazone have been requested to test their products and check their processes to rule out the presence of nitrosamine impurities. More recently nitrosamine impurities have been identified in batches of ranitidine and consequently an EU-wide review has been initiated.
Update [8/01/2020]
Long-term limits of nitrosamines for non-sartan products are still under consideration.
For any new cases of nitrosamine detection in a medicinal product, the MAH should apply, whilst waiting for the outcome of the CHMP Art 5(3) procedure, interim limits calculated for a lifetime treatment and based on a maximum daily dose of the medicine.
Temporary limits for NDMA, NDEA as well as for NMBA, DIPNA, EIPNA and NDBA impurities have been set in line with current ICH M7(R1) guidelines and are given in the table below.
NDMA, NMBA
NDEA, NDBA, DIPNA, EIPNA
Active substance
Maximum daily
dose [mg]
Acceptable Intake [ng/day]
Limit [ppm]
Acceptable Intake [ng/day]
Limit [ppm]
Candesartan
32
96.0
3.000
26.5
0.820
Irbesartan
300
96.0
0.320
26.5
0.088
Losartan
150
96.0
0.640
26.5
0.177
Olmesartan
40
96.0
2.400
26.5
0.663
Valsartan
320
96.0
0.300
26.5
0.082
these limits are not applicable for batches where more than one of the above N-nitrosamines has been identified simultaneously; such batches should be rejected;
NDMA – N-nitrosodimethylamine
NDEA – N-nitrosodiethylamine,
NMBA – 4-(methyl)(nitroso)amino)butanoic acid
NDBA – N-nitrosodibutylamine
DIPNA – N-nitrosodiisopropylamine
EIPNA – N-nitrosoethylisopropylamine
If this interim limit is not exceeded, competent authorities shall be informed on the levels of the impurities detected.
Where the interim limit is exceeded for medicinal products with a limited treatment period or intermittent treatment (e.g. once a week), higher daily exposures may be used as an adjusted interim limit. The approach described in the ICH M7 guideline as the Less Than Lifetime (LTL) approach can be used to calculate adjusted interim limits for impurities present in medicinal products given for LTL and these are described in the following table:
Duration
1 day – 1 month
1 month – 1 year
1 year – 10 years
10 years – lifetime
Daily intake
[ng]
80 x IL
13.3 x IL
6.7 x IL
IL
IL – interim limit
IMPORTANT! Update [28/10/2020]

As a precautionary measure, CHMP does not recommend to generally applying the LTL approach to N-nitrosamine impurities. CHMP agreed that implementation of limits for N-nitrosamines in medicinal products based on ICH M7 principles for substances of the “cohort of concern” and calculated considering a lifetime daily exposure would provide the most appropriate and science-based approach that would also be consistent with the general handling of mutagenic impurities according to ICH M7(R1) and would also allow for the protection of public health. Indeed, this approach is considered sufficiently conservative to protect public health, especially with the additional safe-guard of not allowing a less-than-lifetime approach that could theoretically lead to high acute exposure to nitrosamines from medicinal products used for only short periods of time.
The risk approach is applicable to all routes of administration and no corrections to interim limits are generally warranted unless data justify route-specific differences that should be evaluated case by case.
Nitrosamines are not expected to be formed during the manufacture of the vast majority of APIs outside the class of sartans with a tetrazole ring. However, it is now known that these impurities can form during production under certain conditions and when certain solvents, reagents and other raw materials are used. In addition, impurities can be carried over during the manufacturing process when using already-contaminated equipment or reagents. Furthermore, in cases where nitrosamines can form or are carried over during production, the impurities should normally be controlled and removed during the manufacturing process. Therefore, despite the low risk of nitrosamines being present, Marketing Authorisation Holders (MAHs) are asked to take precautionary measures to mitigate the risk of nitrosamine formation or presence during the manufacture of all medicinal products containing chemically synthesised APIs, i.e. MAHs should review their manufacturing processes to identify and, if found, to mitigate risk of presence of nitrosamine impurities in all human medicinal products containing chemically synthesised active pharmaceutical ingredients.

MAHs should work with manufacturers of API and finished products in order to review the API and finished product manufacturing processes with respect to the arrangements for preventing nitrosamine formation as well as contamination or cross-contamination, taking into account their knowledge of the manufacturing processes as well as the potential sources of nitrosamine impurities.
MAHs are responsible for ensuring that their medicinal products are manufactured in accordance with the requirements laid down in Directive 2001/83/EC. MAHs are responsible for the quality, safety and efficacy of their products, including the quality of the APIs, excipients and raw materials used in the manufacture of finished products. MAHs should therefore ensure (via quality agreements) that they and the holder of the manufacturing authorisation have access to relevant information from the API manufacturers concerning potential formation of nitrosamine impurities and the potential for cross-contamination. The holder of the manufacturing authorisation is also reminded of their responsibility to ensure the use of APIs that have been manufactured in accordance with good manufacturing practice (GMP) for active substances.

The information necessary for risk evaluation should be made available to the MAHs by the manufacturers. Even for products with active substance master files (ASMFs) and certification of suitability to the monographs of the European Pharmacopoeia (CEPs) containing information that is not available to MAHs, MAHs remain responsible for ensuring that robust risk evaluations have been appropriately carried out by the ASMF or CEP holder to enable the MAH to take responsibility for the quality of the active substance and the medicinal product.
EC
Update [20/02/2020]

While the final lesson learned in cases of unexpected nitrosamine impurities in APIs are not available yet, the preliminary draft suggest the necessity to better ensure that marketing authorisation holders (MAHs) fulfil their legal responsibilities for the quality of their product. It is important to strengthen the overview by the MAH taking into account that some of APIs manufacturers concerned did not carry out an investigation of the root cause of the quality issue and did not understand the principles of the quality risk management.
Another point of concern is that in some cases, the MAHs do not have access to enough information regarding the API manufacturing and control process where API is registered via active substance master file (ASMF) or certificate of suitability (CEP), as this information is considered as commercially confidential by API manufacturers.
For medicinal products containing APIs other than sartans with a tetrazole ring,
the following steps should be taken:


Step 1:
Risk evaluation
Step 2:
Confirmatory testing
Step 3: Changes to the marketing authorisation
MAHs should perform risk evaluation of their medicinal products containing chemically synthesised API (i.e. all authorised human medicinal products including generics and over-the counter (OTC) products) using quality risk management principles and evaluate possibility of nitrosamines being present in every concerned medicine at the latest within 6 months of the publication of the notification (26 September 2019),
Update [23/11/2019]
i.e. MAHs should report the outcome by 26 March 2020 at the latest;
Update [26/03/2020]
the deadline to complete step 1 (risk evaluation) is extended to 1 October 2020;
Update [4/09/2020]
for product containing chemically synthesised APIs, the step 1 risk evaluation should be concluded and reported at the latest by 31st March 2021;
for product containing biological APIs, step 1 risk evaluation should be concluded and reported at the latest by 01st July 2021;
Update [05/02/2021]
products that have been approved after September 26, 2019 but for which a risk evaluation was not assessed within the MAA procedure should comply with the call for review deadlines, if not already done so;
Update [8/01/2020]
MAHs of sartans with a tetrazole ring (i.e. those covered by the respective Art.31 referral procedure) should comply with the conditions from the referral; however, considering that additional root causes have emerged, subsequent to conclusion of that referral these additional risk factors should also be considered for products containing sartans with a tetrazole ring;
MAHs should prioritise products in order to establish the sequence in which their products are to be evaluated, starting with medicines more likely to be at risk of containing nitrosamines;
for the purposes of this prioritisation, MAHs may consider factors such as the maximum daily dose taken, duration of treatment, therapeutic indication and number of patients treated; for example, medicinal products with higher daily dose and those for chronic use may take priority;
MAHs should take into account findings from CHMP’s review of sartans;
MAHs should inform the concerned Competent Authorities of outcome of risk evaluations;
IMPORTANT! Update [02/10/2020]
the submission of the risk evaluation template as outcome of step 1 can take place only once; it should therefore be avoided to submit premature risk evaluation outcomes mentioning a potential risk because of missing data;
in the event that a risk of presence of nitrosamines is identified as a result of the risk evaluation, confirmatory testing should be carried out using appropriately validated and sensitive methods in accordance with the prioritisation deriving from the risk evaluation;
Acceptable Intakes (AIs) on which temporary limits should be based for NDMA, NDEA, NMBA, DIPNA and EIPNA are presented in the table above;
products identified as high priority should be tested as soon as possible;
confirmatory testing of all medicinal products identified to be at risk of presence of nitrosamines and submission of required changes in the manufacturing authorisations should be concluded at the latest within 3 years of the publication of the notification (26 September 2019) or at an earlier time if otherwise justified;
Update [7/10/2022]
the CHMP and CMDh extended the deadline for submitting variation applications for chemical medicines from 26 September 2022 to 1 October 2023 in July 2022;
the extension aims to enable companies to perform a thorough investigation and to establish any required risk mitigating actions in light of new scientific developments since 2020, in particular those concerning active substance-derived nitrosamines;
Update [4/09/2020]
for product containing chemically synthesised APIs, confirmatory testing activities at Step 2 and submission of any changes required to Marketing Authorisations (Step 3), are expected to be finalized at the latest by 26th September 2022;
for product containing biological APIs, confirmatory testing activities at Step 2 and submission of any changes required to Marketing Authorisations (Step 3), are expected to be finalized at the latest by 1st July 2023;
MAHs should inform the competent authorities immediately if tests confirm the presence of an nitrosamine impurity irrespective of the amount detected;
MAHs should apply for a variation in a timely manner to introduce any required changes, such as amendment of the manufacturing process or changes to product specifications;
At all steps, timelines should be shortened and authorities immediately informed if findings indicate
an immediate risk to public health.
Update [8/01/2020]
Once steps 1 and 2 are completed, MAHs together with API and finished product manufacturers are expected to maintain the quality of the product throughout its lifecycle and therefore to review the outcome of the risk evaluation and testing as and when new information on potential root causes for nitrosamine formation or contamination becomes available. Appropriate timelines for conducting the risk evaluation and testing for the newly identified risks should be implemented depending on the level and impact of the risk identified.
NEW! Update [25/10/2022]
Active substance-derived nitrosamines
Authorities in the EU are aware that some active substances are at a higher risk of formation of active substance derived nitrosamine impurities.
A particular risk of formation of nitrosamines should be noted for active substances that contain a nitrosatable amine functional group. Several examples have been reported where the amine functionality was shown to be vulnerable to nitrosation and formation of the corresponding N-nitroso impurity (i.e. NO-API). Active substances that contain secondary amines appear particularly vulnerable to this reaction, although some cases involving active substances with tertiary amines have also been observed. Vulnerable amines could also be formed by degradation (e.g. hydrolysis) during formulation or storage. Nitrites have been identified as impurities in many common excipients. MAHs and/or applicants should be aware that N-nitroso API impurities can form at levels exceeding the AI even if nitrite levels in the excipients are very low.
EDQM
Update [23/11/2019]
Although EDQM does not expect that the issue regarding presence of nitrosamines impacts many substances, it is now appropriate to expand the review to all other APIs (other than sartans with a tetrazole ring). manufactured from chemical synthesis for which CEPs have been granted. EDQM therefore requests that the holders of such CEPs follow the process described here (which is a similar stepwise approach to that for MAH).
Step 1:
Risk evaluation
Step 2:
Confirmatory testing
Step 3:
Revision to the CEP
companies holding CEPs should perform a risk evaluation of their chemically synthesised APIs with regards nitrosamine formation, using quality risk management principles, as outlined in the ICH Q9 guideline;
the CEP holders should prioritise substances in order to establish the sequence in which they are to be evaluated;
if any substances are identified as high priority, the risk evaluation should be done immediately;
the risk evaluation should address not only risks from the manufacturing process but also those from the introduction of materials used in the manufacturing process (e.g. starting materials, reagents, solvents – fresh and recovered etc.);
the risk evaluation for all CEPs should be concluded at the latest by 26th March 2020;
Update
the deadline to complete step 1 (risk evaluation) is extended to 31 July 2020 due to the COVID-19 pandemic;
CEP holders are not required to confirm to EDQM that step 1 has been completed where no risk is identified; however where a risk is identified, EDQM should be immediately informed and the company should progress to step 2, confirmatory testing, and the timescales for when the confirmatory testing results will be provided to EDQM should be indicated to EDQM;
the outcome of step 1 relating to the risk evaluation performed for a CEP should be communicated to the customers in all cases (even if no risk is identified) such that the MAHs can use this information to fulfil their responsibilities;
in the event that a risk of presence of nitrosamines is identified as a result of the risk evaluation, confirmatory testing should be carried out using appropriately validated and sensitive methods in accordance with the prioritisation deriving from the risk evaluation conducted in step 1;
substances identified as high priority should be tested as soon as possible;
all APIs identified to be at risk of presence of nitrosamines should be tested and the results provided to EDQM with, if needed, a proposal for subsequent actions (such as revision of the CEP);
CEP holders should inform the EDQM immediately if the tests confirm the presence of a nitrosamine impurity, irrespective of the amount detected, and provide the results;. the companies should then progress to step 3 after informing EDQM of the plan and timescales to complete step 3;
where nitrosamine impurities have been detected, CEP holders should apply for a revision to their application(s) in a timely manner to introduce any required changes, such as amendment of the manufacturing process or changes to specifications and introduction of controls;
the required revisions to the CEP applications should be concluded at the latest by 26th September 2022 or at an earlier time if otherwise justified;
CEP holders are reminded that it is their responsibility to complete the procedure described above for all their impacted CEPs.
EDQM may contact CEP holders for information on the risk of nitrosamines at any step of this review and any such requests should be responded to fully and in a timely manner.
EDQM will take action on any CEP (e.g. suspension) where information becomes available regarding an unacceptable level of nitrosamine impurities in the active substance which is the subject of a CEP.
EDQM reminds CEP holders that they should provide the appropriate information relating to the risk evaluation they have performed for their CEP to their customers in all cases (for steps 1 to 3, and for step 1 even if no risk has been identified) such that the MAHs can use this information to fulfil their responsibilities in a timely manner.
Update [16/09/2020]

It is important to note that from 1 October 2020, applicants and CEP holders should systematically include a risk assessment regarding the potential formation of nitrosamines in any new CEP application, sister file or renewal application, as well as in any revision where a risk of nitrosamine formation may be introduced (i.e. changes to the manufacturing process, change of suppliers of starting materials or intermediates, etc.).
Source: EMA, EDQM, EC
Related entries: